Overview
General description
MetaNetMap is a Python tool dedicated to mapping metabolite information between metabolomic data and metabolic networks. The goal is to facilitate the identification of metabolites from metabolomic data that are present in one or more metabolic networks to facilitate further modelling, taking into consideration that data from the former likely has distinct identifiers from the latter.
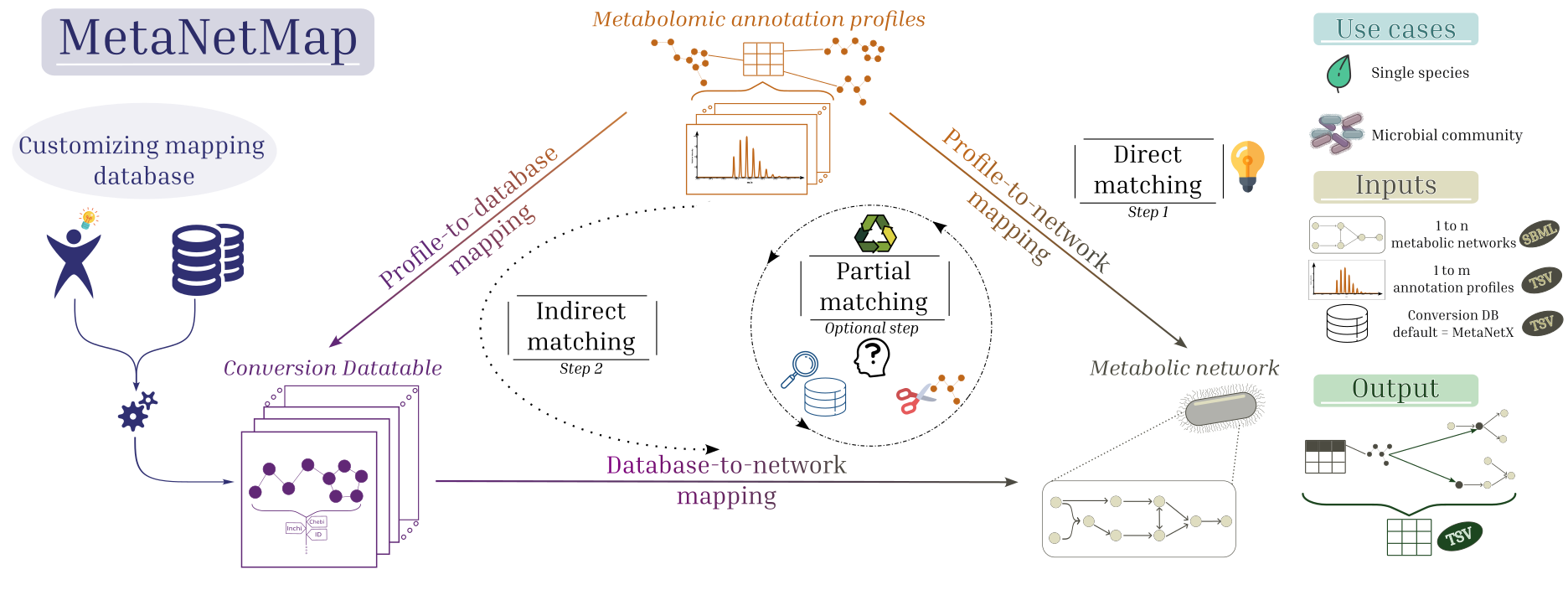
While some metabolites can be matched directly using common identifiers, many require more sophisticated approaches. MetaNetMap implements such strategies to improve mapping rates, including optional partial matching techniques. The picture above summarises the mapping procedure implemented in MetaNetMap.
Why using this tool to map metabolomic data?
Mapping metabolomic data to metabolic networks is a challenging task for several reasons:
Identifier variability in metabolic networks: Automatic reconstruction of metabolic networks using different tools and associated databases often assigns distinct identifiers to the same metabolites. It is likely that those do not match the nomenclature of metabolomic annotations. To reconcile them, a first solution is to rely on metadata extracted from metabolic networks associating molecules to alternative databases. Additionally, third-party external databases such as MetaNetX can be used, in order to provide more matching possibilities.
Metabolomic data complexity: Due to the difficulty of annotating metabolomic profiles, identifications are often partial, incomplete, and inconsistently represented. For example, enantiomers are frequently not precisely specified because they are almost indistinguishable by classical LC/MS methods.
While a few metabolites can be matched manually with limited effort, large-scale metabolomic datasets require automated tools to perform mapping efficiently and accurately. MetaNetMap addresses these challenges by providing a robust framework for mapping metabolomic data to metabolic networks, leveraging multiple strategies to maximize matching success. In practice, MetaNetMap can match one or several metabolomic annotation tables to one or several metabolic networks.
Third-party database for matching
In case metadata from metabolic network do not match identifiers of the metabolomic data, a conversion_datatable file acts as a bridge between the metabolomic data and the metabolic networks.
MetaNetMap enables the construction of such resource using MetaNetX or MetaCyc knowledge bases. In the former case, data from chem_xref.tsv and chem_prop.tsv MetaNetX files is used. In the latter case (requires a licence), metadata from the compounds.dat file is extracted. Additionally, users can provide another table with existing mapping data resulting from previous curation efforts for instance, referred to as datatable_complementary.
The conversion table serves as a comprehensive knowledge base that allows MetaNetMap to search across all known identifiers for a given metabolite and match them between the input data and the metabolic networks.
Refer to the related documentation Usage to build your first mapping table using MetaNetX data.
Note
The test commands of MetaNetMap rely on MetaCyc database.
However, complete information from MetaCyc related to the ontology of metabolites and pathways is not included in the test option because of licensing restrictions.
Only a simplified example (a “toy” version) of the conversion_datatable file is provided. If you have a MetaCyc licence, you can build the complete conversion table using the compounds.dat file from MetaCyc database.
After building this knowledge base, mapping can be performed in two modes:
Classic mode: The classic mode allows you to input one metabolomic data file or a directory containing several of them, and a unique metabolic network.
Community mode: The “community” mode allows you to input a directory containing one or several metabolomic data files, as well as a directory containing multiple metabolic networks. It will map each metabolomic data file against each metabolic network file, resulting in a comprehensive mapping across all combinations. This mode is useful for large-scale analyses involving a microbial community where multiple organisms and their associated networks are considered in the metabolomic study.
▸ Partial match (Option for mode classic and community): The partial match is a post-processing step applied to metabolites or IDs that were not successfully mapped during the initial run. These unmatched entries are re-evaluated using specific strategies, which increase the chances of finding a match (e.g., via ChEBI, InChIKey, or enantiomer simplification). This step is optional, as it can be time-consuming depending on the number of unmatched entries.
Overview of the procedure
Mapping procedure
Step 1: Match metabolomic data vs. metabolic network metadata
We first test for direct matches between the metabolite information in the metabolomic data and all the metadata available in the metabolic networks, without initially using the
conversion_datatable. For each metabolite that matches, we then look up the matched identifier in theconversion_datatableto retrieve its unique ID within this database. This step will later allow us to detect potential ambiguities with other matches during the indirect matching and to confirm its presence in the database.Step 2: Match metabolomic data vs. conversion_datatable
Metabolites from metabolomics that did not match in the previous step will be tested here. Duplicate checks will be performed, since multiple columns from the metabolomic inputs will be tested for the same metabolite (i.e., within a single row). It is therefore possible that several identifiers of the conversion table match the same metabolite. In this case, the matches will be merged in the output table, separated by
_AND_. For example, the CPD-17381 and roquefortine C are identifiers that correspond to the same metabolite. In this case, it will be written as CPD-17381 _AND_ roquefortine C.Only matches are provided as outputs. Non-matching identifiers for a given metabolite will be excluded from MetaNetMap outputs to improve readability.
Step 3: Match metabolites in conversion_datatable vs. metabolic network metadata
This step continues the matching process for the metabolites that were mapped on the conversion datatable at Step 2, in order to associate them to metabolic network information, considering the additional identifiers that this knowledge base contains. Step 3 therefore search for matches against any metabolic network metadata.
If none of the metabolic network identifiers match any conversion table reference, the information that a match with the conversion table occurred will still be provided in the output. If several distinct matches occurred, all of them will be merged in the result file (separated by
_AND_), as they represent the same metabolite. This allows all information for one metabolite to be grouped on a single row, improving clarity and readability.
For more details on advanced methods (partial match, ambiguities, …), see: Advanced usage
License
GNU Lesser General Public License v3 (LGPLv3)